Description
PrediSweet predicts the sweetness of a molecule from its SMILES (Simplified Molecular Input Line Entry Specification). The complete modeling and prediction procedure is written in Python, and uses the following packages:
- Scikit learn

- RDKit

- Mordred

- ChemoPy

- Standardiser

- SciPy

Usage
-
Input
3 options are available:- Drawing: generated by Ketcher. The resulting molecule is then automatically converted to the SMILES format
- Text: directly input one or more SMILES, separated by a newline
- File: containing 1 SMILES per line
-
Output
- Name: if possible, the name displayed in PubChem, else the Inchi-key of the molecule
- Molecule: a 2D drawing of your molecule
- Status: current step of the sweetness prediction protocol
- log Sweetness: the relative sweetness of your molecule, in log10 scale. Sucrose is used as a reference.
- Applicability Domain: indicates if the query compound is within the descriptors range of the training set. If the score is not 1, the model is not applicable to the query compound, thus the prediction should be discarded.
- Reliability Domain: indicates the density of information around the query compound. If the score is below 1, not many training set compounds are present in the vicinity of the query, thus the prediction is expected to be poorly reliable.
- Decidability Domain: indicates the confidence in the prediction that was made, based on the intermediate conclusions given by the model. If the score is below 0.5, the prediction is not supported by enough evidence. This score provides valuable information for risk assessment and should guide the decision process (provided the query compound is in the applicability and reliability domains).
The color code on the 3 domains of applicability is here to summarize the following information:- Red if the query is outside of the applicability domain: the model is not applicable in this case
- Orange if the query is in the applicability domain, but outside of the reliability domain: the model is not reliable in this case
- Yellow if the query is in the applicability and reliability domains, but outside the decidability domain: the model is applicable and reliable for the query, but the prediction is disputed
- Green if the query is within all domains: the model is applicable and reliable for the query, and the prediction is certain
Workflow and implementation
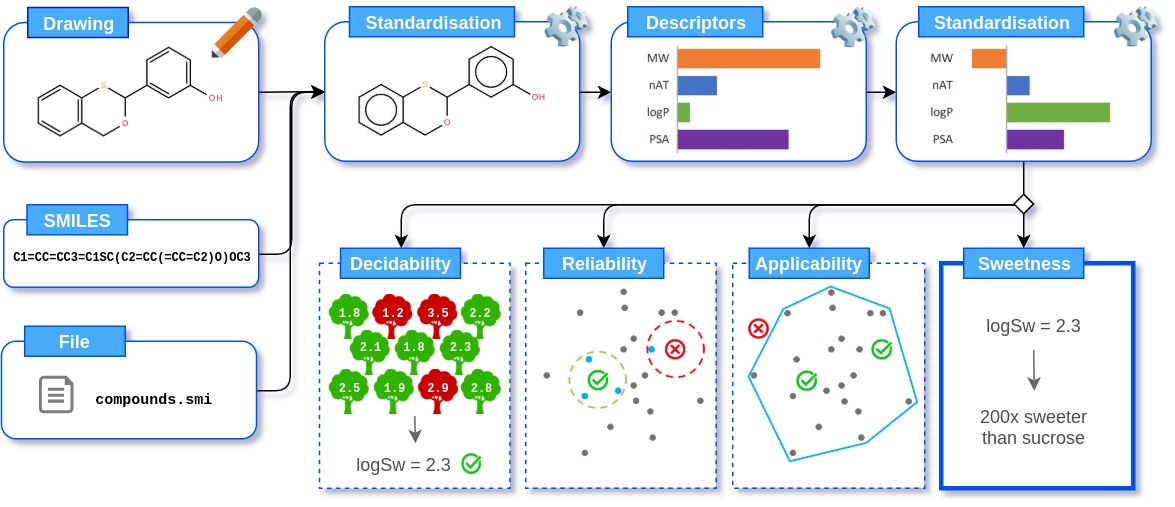
- Generate the molecule from SMILES, and retrieve its name: RDKit, and pubchempy
- Remove salts and Standardise the structure: RDKit, and standardiser
- Calculate and standardise 0D, 1D and 2D descriptors: Mordred (37 descriptors), RDKit (6 descriptors), and ChemoPy (8 descriptors)
-
Predict the Sweetness: performed using the Adaptive Boosting (AdaBoost) algorithm on a set of 80 Decision Trees, as implemented in the scikit learn package.
The resulting value is in log10 scale and represents the concentration ratio between a sucrose solution and a solution of sweetener perceived with the same intensity. For example, if a 1 mM solution of sweetener is perceived with the same intensity as a 10 mM solution of sucrose, its sweetness is 10 and the logSweetness value is 1. -
Estimate the Applicability score: a Convex Hull approach is used. Because of the high dimensionality, the hull is not computed on the 51 descriptors used by the model, but on the 6 principal components of a PCA fitted on the training set, and accounting for 89% of the variance.
The resulting hull is made of 103 vertices and 4 754 facets. It is slightly expanded by moving each vertex lightly away from the centroid of the hull.
Every facet of the resulting hull can be described by an equation consisting of a normal vector V and an offset b. If a query point 'x' is inside the hull, then it must satisfy the inequality 'V.x + b < 0' for every facet.
The applicability score returned by the server represents the number of inequalities that were satisfied, divided by the number of facets. -
Estimate the Reliability score: a fixed-radius near neighbors approach is used. A hypersphere of radius 7.0 in the descriptor space is drawn around the query compound, and the number of training set compounds that lies within the sphere is counted. If the count is superior or equal to a threshold, the query is defined as reliable.
The reliability score returned by the server is the number of neighbors in the sphere divided by the threshold (3 near neighbors for a radius of 7.0). -
Estimate the Decidability score: based on the predictions of each individual Decision Tree of the AdaBoost model. In AdaBoost, each weak learner (here, a Decision Tree) is associated with a weight that describes its accuracy when it was trained.
To compute the decidability score, the final prediction is compared to the prediction of each individual tree. The score is computed as the sum of the weights of trees that made a prediction equal to that of the model ± ɛ, over the sum of all weights. Here, ɛ = 0.5, which is close to the mean absolute error of the model on the test set.
Known errors
- Failed reading: RDKit could not generate a valid molecule from the input SMILES
- Failed standardisation: the input molecule was treated as a salt by standardiser (and was removed), or its structure could not be standardised
- Failed descriptors calculation: a descriptor used by the model could not be computed
- Failure: timed out: the input molecule took to long to be predicted and the calculation was killed by the server
Please report any issue on our GitHub page